Last updated: May 01, 2013

2013 Release Tcga Researchers Identify Potential Drug Targets Markers For Leukemia Risk

TCGA researchers identify potential drug targets, markers for leukemia risk
New study reveals relatively few mutations in AML genomes
 |
The findings - which appeared online May 1, 2013 - in the New England Journal of Medicine set the stage for identifying potential new drug targets and treatment strategies for AML. They may also offer better guidance for predicting the severity of disease for individual patients.
"These results provide important new insights into the genomics of a deadly and difficult-to-treat cancer, and underscore the power and scope of The Cancer Genome Atlas project," said NIH Director Francis S. Collins, M.D., Ph.D.
"Rather than just random snapshots about individual patients, this study provides a more detailed look at the aberrant genomes of AML than we have ever had before," said NHGRI Director Eric D. Green, M.D., Ph.D. "It has the potential to open up new directions in AML research, and perhaps, in the design of new therapeutics, its impact could be felt in the near future."
AML, the most common acute form of adult leukemia, develops when immature white blood cells fail to mature and instead accumulate in the bone marrow. The leukemia cells reduce the production of healthy blood cells, leading to anemia, abnormal bleeding and infections, and, if untreated, death.
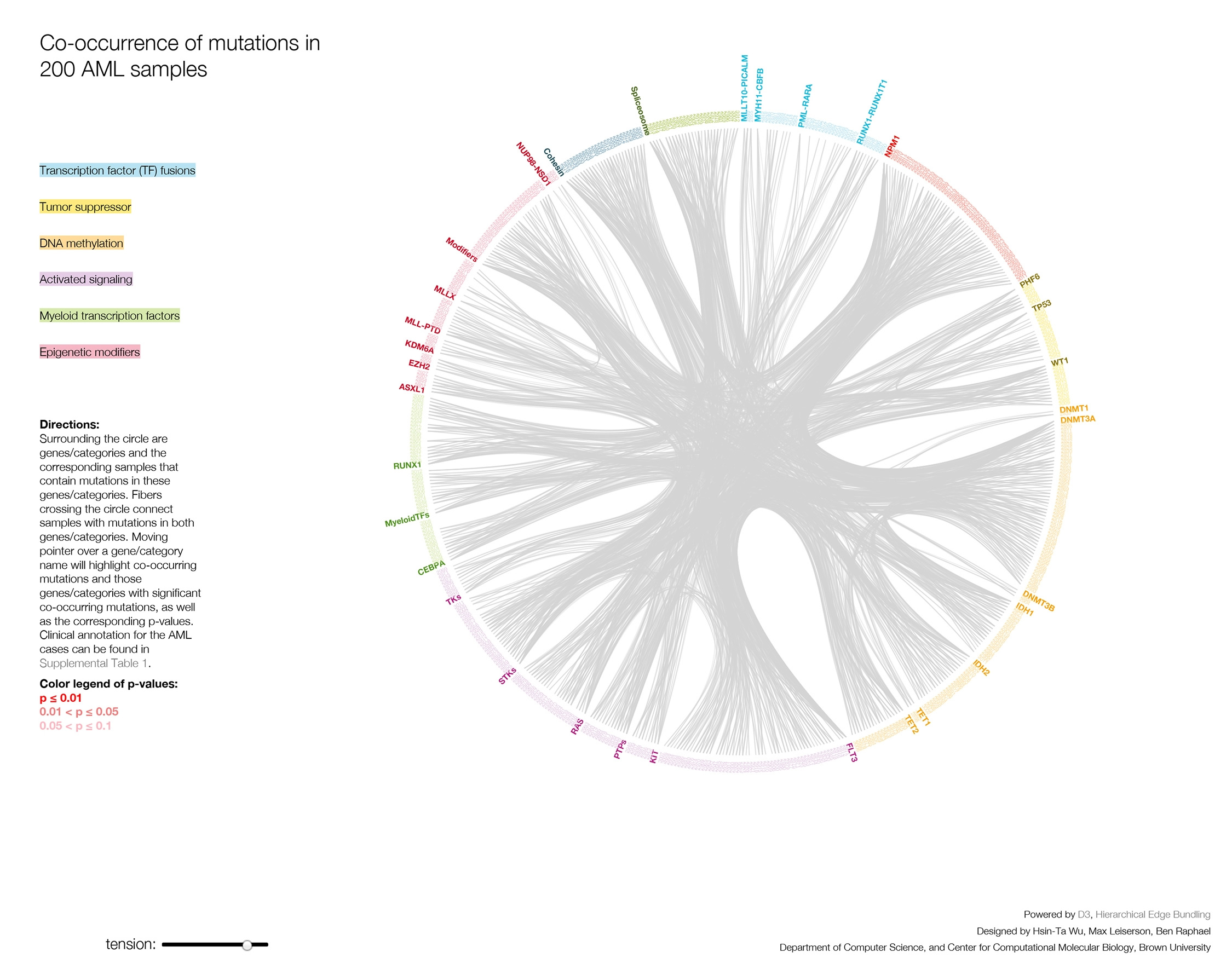
Researchers examined the genomes of tumor specimens from 200 adult cases of spontaneously occurring, newly diagnosed AML. These cases represented all of the known subtypes of AML in approximately the same proportion as the general population. In this way, the study provided a realistic view of the disease, particularly in the number and frequency of genomic alterations. Each AML genome was compared to the normal genome derived from a skin sample of the same patient. Out of the 200 samples, 50 were analyzed by using whole genome sequencing, which is an examination of the complete DNA blueprint of the cells. Researchers analyzed the genome's protein-coding regions in the remaining samples, and used powerful new sequencing methods to look at changes in RNA in each case.
By studying a large number of AML cases, investigators were able to predict that they have identified virtually all of the mutations that occur in at least 5 percent of AML patients. Surprisingly, they found that overall, AML genomes have relatively few mutations, and such tumors are among the least mutated adult cancers. The average number of mutations in genes for each AML genome was 13, in contrast to solid tumors such as breast, lung or pancreatic cancer, which often have hundreds of mutated genes.
Because investigators found more than 1,600 genes that were mutated at least once in the 200 samples, they organized the recurrently mutated genes into nine categories based on their function or the known pathways involved. Some of these categories include tumor suppressor genes, signaling genes and epigenetic modifiers, with the latter being the most frequently mutated class of genes in the study. Epigenetic changes are alterations to DNA that often involve the addition or removal of chemical tags (such as methyl groups), which can affect when genes are turned on and off.
"This data set helps to integrate what was previously fragmented information," said study co-leader Timothy J. Ley, M.D., associate director for cancer genomics at The Genome Institute at Washington University School of Medicine in St. Louis. "We didn't realize how few recurrent mutations there were, and no one was thinking even five years ago that AML was associated with a high frequency of mutations in genes that encode epigenetic modifiers."
By finding comparatively few recurrently mutated genes, yet frequent alterations in genes that help control gene expression, the investigators may have narrowed the search for likely drug targets and disease markers.
Other results were also somewhat surprising. Researchers knew that mutations in signaling genes, which help control cell growth and development, were very common in AML, and thought that all AML samples may have at least one signaling gene mutation. But the TCGA findings showed that these genes are mutated in only 60 percent of cases. These include mutations in the gene FLT3, which occur in about a third of cases, making it one of the most commonly mutated genes in AML. FLT3 is important for normal blood cell development. The researchers also found that many AML patients have concurrent mutations in three commonly mutated genes: FLT3, NPM1 and DNMT3A. Patients with this combination of gene mutations appear to have a unique subtype of AML.
Investigators unexpectedly found recurring mutations in cohesin genes, which are important in cell division.
The study is the first to report a recurrently mutated microRNA gene in AML. MicroRNAs can play an important role in regulating gene expression, particularly in turning off gene activity.
Abnormal chromosome rearrangements and gene fusions (where two genes join to form a new, altered gene) are frequently useful in diagnosing and providing prognostic information for AML patients. The study uncovered many such fusions that had not been described before, and nearly half of the AML samples were found to have gene fusions.
Currently, only a few good markers exist to help guide treatment decisions for the majority of patients with intermediate risk. Some of the recurrently mutated genes identified in this study may allow for better prognostic information that will be relevant for AML patients.
"We've never had such a complete picture of AML, and this data set will be mined by researchers for years," said co-study leader Richard Wilson, Ph.D., director of Washington University's Genome Institute. "These findings have probably identified every pathway in which a modification - and perhaps new drugs - might be beneficial. They also further refine our understanding of the importance of individual mutations for disease classification and prognostication, and will help us build better disease models."
"These results will enable investigators to examine patient samples for mutation patterns and affected pathways, and to begin new studies to try to understand the relationships between these genetic mutations and treatment results," added Ley.
"This study of AML reinforces the value of the approach we are using to study the genomic diversity among tumors of many different cancer types and even within a single kind of cancer such as AML," noted NCI Director Harold Varmus, M.D. "Only such a systematic analysis of cancer types could have uncovered such clear patterns, such as the apparent importance in AML of genetic mutations that lead to changes in gene expression and cell traits."
To date, TCGA Research Network has published analyses on these cancers:
- glioblastoma multiforme
- ovarian serous adenocarcinoma
- colorectal adenocarcinoma
- lung squamous cell carcinoma
- invasive breast cancer
This work was supported by the following grants: U24CA143845, U24CA143858, U24CA144025, U24CA143882, U24CA143866, U24CA143867, U24CA143848, U24CA143840, U24CA143835, U24CA143799, U24CA143883, U24CA143843, U54HG003067, U54HG003079, U54HG003273, and P01CA101937.
Reference: The Cancer Genome Atlas Network. Genomic and epigenomic landscape of adult de novo acute myeloid leukemia. New England Journal of Medicine. Online May 1, 2013. In print May 30, 2013. DOI: 10.1056/NEJMoa1301689.
The TCGA Research Network consists of more than 150 researchers at dozens of institutions across the nation. A list of participants is available at http://cancergenome.nih.gov/abouttcga/overview. More details about The Cancer Genome Atlas, including Quick Facts, Q&A, graphics, glossary, a brief guide to genomics and a media library of available images can be found at http://cancergenome.nih.gov.
NHGRI is one of the 27 institutes and centers at the National Institutes of Health. The NHGRI Extramural Research Program supports grants for research and training and career development at sites nationwide. Additional information about NHGRI can be found at http://www.genome.gov.
NCI leads the National Cancer Program and the NIH effort to dramatically reduce the burden of cancer and improve the lives of cancer patients and their families, through research into prevention and cancer biology, the development of new interventions, and the training and mentoring of new researchers. For more information about cancer, please visit the NCI website at http://www.cancer.gov or call NCI's Cancer Information Service at 1-800-4-CANCER (1-800-422-6237).
About the National Institutes of Health (NIH): NIH, the nation's medical research agency, includes 27 institutes and centers and is a component of the U.S. Department of Health and Human Services. NIH is the primary federal agency conducting and supporting basic, clinical, and translational medical research, and is investigating the causes, treatments, and cures for both common and rare diseases. For more information about NIH and its programs, visit http://www.nih.gov.
Contact:
NHGRI Communications
(301) 402-0911
mccrimmono@mail.nih.gov
NCI Press Office
(301) 496-6641
ncipressofficers@mail.nih.gov
Posted: May 1, 2013