
Regulación de pruebas genéticas
A medida que la ciencia de la genómica avanza, las pruebas genéticas se están convirtiendo en algo más común en la práctica clínica. Aún así, la mayoría de las pruebas genéticas no son reguladas, lo cual significa que están disponibles en el mercado sin ningún análisis independiente para verificar las menciones expresas del vendedor sobre las pruebas.
-
Regulación federal de pruebas genéticas
Tres agencias federales desempeñan una función en la regulación de las pruebas genéticas: el CMS, la FDA y la Comisión Federal de Comercio (Federal Trade Commission, FTC).
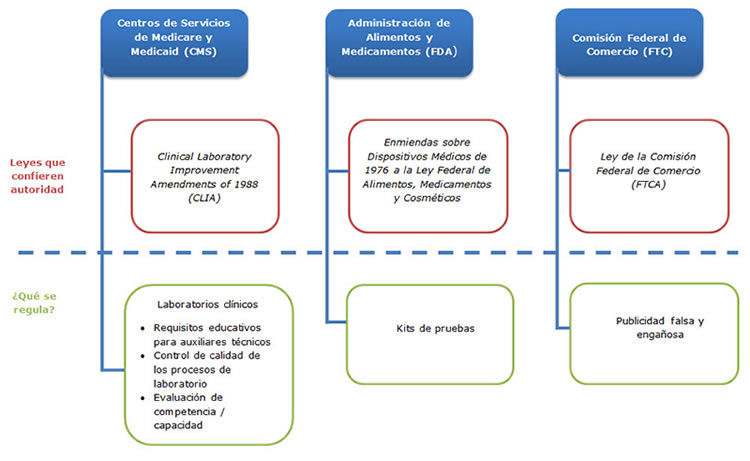
El CMS es responsable de regular todos los laboratorios clínicos que hacen pruebas genéticas, para asegurar su cumplimiento con las Enmiendas para Mejoras de Laboratorios Clínicos (Clinical Laboratory Improvements Amendments, CLIA) de 1988. El objetivo de CLIA es certificar la calidad de las pruebas clínicas, incluida la verificación de los procedimientos utilizados y la capacidad de los técnicos que procesan las pruebas. También abarca la evaluación de competencia / capacidad para algunas pruebas.
Cómo se evalúa una prueba genética:
- ¿Es la prueba exacta y fiable? (Validez analítica)
- ¿Es médicamente significativo el resultado de la prueba? (Validez clínica)
- ¿Mejora la prueba la atención de la salud? (Utilidad clínica)
La FDA posee la autoridad más extensa en términos de regulación de la seguridad y eficacia de las pruebas genéticas como dispositivos médicos bajo la Ley Federal de Alimentos, Medicamentos y Cosméticos (Federal Food, Drug, and Cosmetic Act). El hecho de que la FDA regule o no una prueba se determina mediante cómo se introduce la prueba al mercado. Una prueba puede comercializarse como un "kit" de pruebas comercial, un grupo de reactivos utilizados en el procesamiento de muestras genéticas que se empaquetan juntos y se venden a múltiples laboratorios. Más comúnmente, una prueba se introduce al mercado como una prueba desarrollada por un laboratorio (laboratory-developed test, LDT); en tal caso, la prueba es desarrollada y realizada por un solo laboratorio, y las muestras se envían a ese laboratorio para ser analizadas. La FDA regula solamente las pruebas vendidas como kits y, a la fecha, ha practicado "discreción en la aplicación" para las LDT.El grado de supervisión de la FDA de una prueba genética se basa en su fin previsto y en los riesgos que representa un resultado inexacto de la prueba. La FDA clasifica los dispositivos médicos, incluidas las pruebas genéticas, en tres clases separadas, que varían desde la clase I, para los productos de riesgo relativamente bajo, hasta la clase III, donde las pruebas se someten al nivel más alto de escrutinio. Puede encontrarse aquí una lista completa de pruebas genéticas humanas aprobadas.
La supervisión por parte de la FDA también incluye la farmacogenómica, que es el uso de información genómica para ayudar a predecir cómo pudiera un individuo responder a un medicamento específico, para identificar individuos que pudieran tener una reacción adversa al tomar un medicamento o para ayudar en la selección de la dosis óptima de un medicamento. Parte de la supervisión por parte de la FDA de los medicamentos comercializados es para asegurar que los fabricantes presenten información en etiquetas de medicamentos acerca de marcadores genéticos, que sea pertinente para la seguridad y eficacia del medicamento. Puede encontrarse aquí una lista de medicamentos farmacogenómicos aprobados.
En comparación con la FDA y el CMS, la autoridad regulatoria de la FTC es relativamente estrecha, y se limita a cómo se anuncian las pruebas. La Comisión tiene la autoridad de regular la publicidad que comunica información relacionada con la salud a los consumidores para asegurar que no sea falsa o engañosa.
Last updated: December 31, 1969